The Great London:
Genetics
Natural Heritage: Sampling species' DNA trails is leading to better environmental monitoring

Evolution: Sex cells evolved to pass on quality mitochondria

Genetics: Obesity in humans linked to fat gene in prehistoric apes
Genetics: Scientists sequence ancient British 'gladiator' genomes from Roman York

Evolution: Rooting the family tree of placental mammals

Genetics: DNA analysis reveals Roman London was a multi-ethnic melting pot

Forensics: Slavery carried bilharzia parasites from West Africa to the Caribbean

Genetics: Mummies from Hungary reveal TB's Roman lineage

Breaking News: Complex genetic ancestry of Americans uncovered

Genetics: Genes for nose shape found

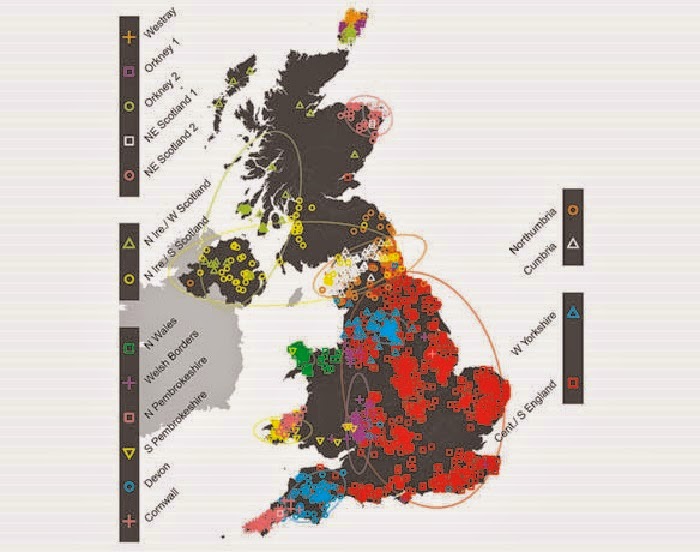
Genetics: First fine-scale genetic map of the British Isles
Oceans: Rising carbon dioxide levels stunt sea shell growth

Genetics: A 100-million-year partnership on the brink of extinction

Near East: Face of 9,500 year old Neolithic man from Jericho reconstructed

Evolution: Scientists reconstruct largest ever family tree of major flowering plant group

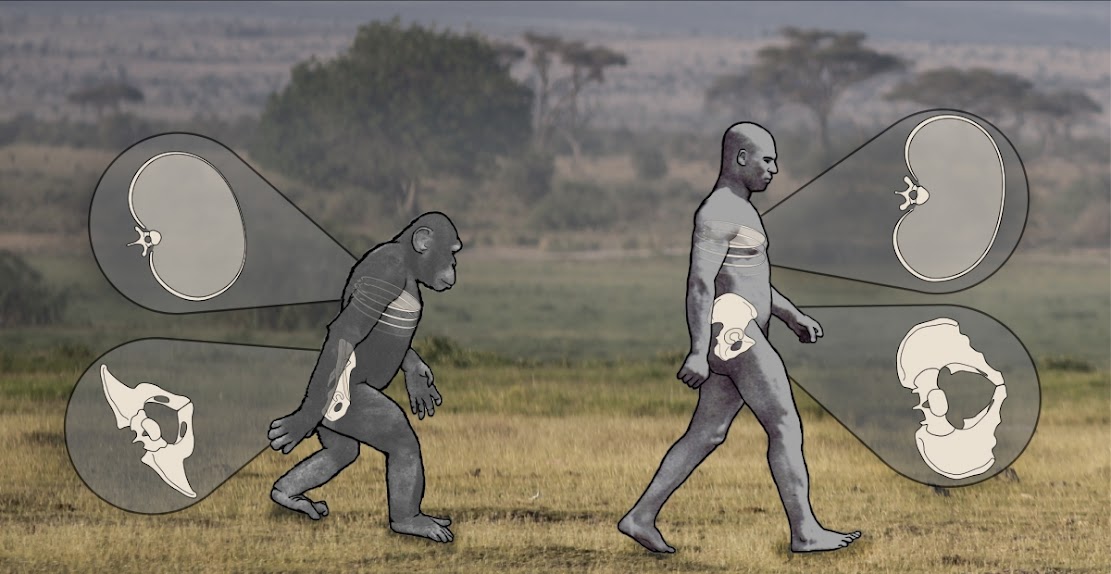
Genetics: Tweak in gene expression may have helped humans walk upright

Polynesia: Forensic analysis of pigtails to help identify original 'mutineers of H.M.S. Bounty'

Genetics: Scientists propose new evolution model for tropical rainforests

Evolution: Life as we know it most likely arose via 'long, slow dance'

Evolution: Divergent climate tolerances play crucial roles in how species evolve
