The Great London:
Genetics
Natural Heritage: Sampling species' DNA trails is leading to better environmental monitoring

Evolution: Sex cells evolved to pass on quality mitochondria

Genetics: Obesity in humans linked to fat gene in prehistoric apes
Genetics: Scientists sequence ancient British 'gladiator' genomes from Roman York

Evolution: Rooting the family tree of placental mammals

Genetics: DNA analysis reveals Roman London was a multi-ethnic melting pot

Forensics: Slavery carried bilharzia parasites from West Africa to the Caribbean

Genetics: Mummies from Hungary reveal TB's Roman lineage

Breaking News: Complex genetic ancestry of Americans uncovered

Genetics: Genes for nose shape found

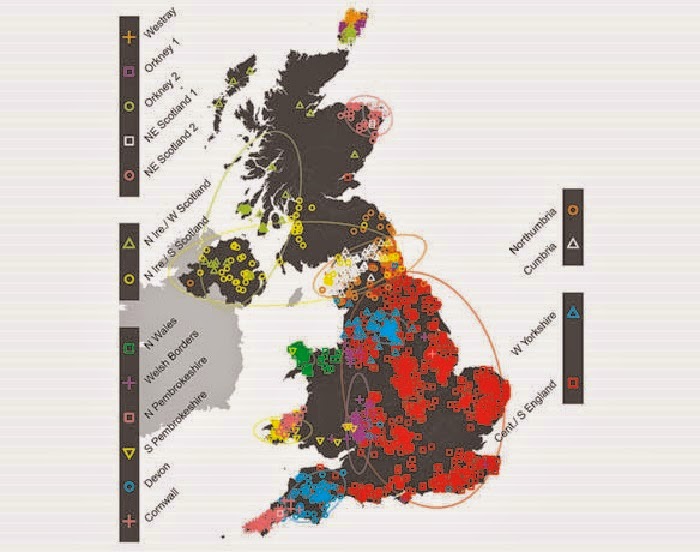
Genetics: First fine-scale genetic map of the British Isles